
Abb. 1: Scolopendra cingulata

Abb. 2: Lithobius forficatus.
| Adulte Scolopendra cingulata
(Abb. 1) wurden im April 1991 im Norden Spaniens
lebend gefangen. Im Labor wurden die Tiere bei Raumtemperatur (20-25°C)
einzeln in Bellaplast-Schalen (10 × 13 × 4 cm3) gehalten,
deren Boden etwa 1-2 cm hoch mit sandigem Kies bedeckt war. Als Unterschlupf
wurden Stücke von Papierhandtüchern und Schieferplättchen
in die Schalen gelegt. Der Boden wurde nach Bedarf mit Leitungswasser befeuchtet.
Einmal pro Woche wurden die Tiere mit Larven oder Imagines von Mehlkäfern,
Fliegen oder Heimchen gefüttert
|
Lithobius forficatus (Abb. 2) wurde
in der Umgebung von Gießen, Leihgestern (Hessen) und Aschaffenburg
(Bayern), gesammelt. Sie wurden ebenfalls einzeln in Bellaplast-Schalen (6
× 4 × 4 cm3) bei Raumtemperatur gehalten, allerdings
auf stets feuchtem, lehmig-humösen Boden. Gefüttert wurde sie mit
flügellosen Drosophila sp. und Stücken von Mehlwürmern
oder Kellerasseln.
|
| Vor der Entnahme von Haemolymphe wurden die Tiere in ihren
Schälchen mit CO2 narkotisiert und auf einer Moosgummiunterlage
mit Parafilm® (American CanCo) festgelegt. Eines der hinteren
Beine wurde im Bereich der Tibia abgeschnitten und die austretende Haemolymphe
entweder direkt im Versuch benutzt oder in kalibrierten (bei Bedarf auch
silikonisierten) Einweg-Glaskapillaren
(VITREX) aufgenommen, um das Volumen zu bestimmen. Die Haemolymphe mehrerer
Tiere wurde in eisgekühlten Eppendorf-Reaktionsgefäßen gesammelt
und tiefgefroren.
Die Gesamthaemocytenzahl wurde mit einer Bürker-Zählkammer von 0,02 mm Tiefe auf den Quadraten mit 0,2 mm Kantenlänge in unverdünnter |
Haemolymphe ausgezählt, die Osmolarität der Haemolymphe mit
einem Osmometer (OM 801, Vogel GmbH) in jeweils 20 µl Haemolympheproben
gemessen.
Eine Abschätzung des pH-Werts erfolgte auf pH-Papier Neutalit® pH 5,5-9,0 (Merck). Mit einem Bürolocher daraus gestanzte Plättchen wurden auf einem Objekträger mit 10 µl a. demin. befeuchtet und ihnen gleich 10 µl Haemolymphe zupipettiert. Nach einer min konnte durch Farbvergleich der pH-Wert abgelesen werden. |
| Für die kurzfristige Haemocytenkultur diente Graces-Medium (GRACE 1962). In einigen Versuchen wurden 'WENNING-Ringer' (artifizielle Lithobius-Haemolymphe nach WENNING et al. 1991) oder Kalium-Ringer (K-Ringer) als Medium benutzt (Tab. 1). | Die Medien wurden durch Einmal-Filterhalter mit 0,2 µm Porengröße (FP030/3, Schleicher & Schuell) in zuvor autoklavierte Gefäße sterilfiltriert und verschlossen. Gekühlt konnten die Medien bis zu zwei Wochen aufbewahrt und benutzt werden. |
| Für die Lichtmikroskopie (LM) wurden Glasobjekträger
oder Deckgläschen mit heißem Wasser und anschließend mit
a. demin. gespült und im Preßluftstrom getrocknet. Aufwendigere
Reinigung (Waschen in RBS oder Ethanol) und Beschichtung mit Poly-L-Lysin
sind nicht vorteilhaft für die Herstellung von
Spreitungspräparaten.
Auf die gereinigten Objekträger kamen etwa 200 µl Kulturmedium zu dem die Haemolymphe direkt vom Tier getropft wurde. Zur Verteilung der Haemocyten wurde mit einer zugeschmolzenen Pasteurpipette kurz gerührt. In der Feuchten Kammer (rundes Filterpapier in Petrischalen, = 16 cm, mit a. demin. befeuchtet) konnten sich die Haemocyten innerhalb von 15-60 min spreiten. |
Für Transmissionselektronenmikroskopie (TEM) und
Rasterelektronenmikroskopie (REM) wurden Haemocyten auf Cellophanfolie
gespreitet. Die Cellophanstücke wurden zuvor in a. demin.
gespült und autokalviert, in Kulturmedium für 30 min equilibriert,
1 × 1 cm2 der Folie in Glasblockschälchen dann mit
Kulturmedium überschichtet und Haemolymphe dazugegeben. Die so erhaltenen
Spreitungspräparate wurden nach der Fixierung in mehrere Stücke
zerteilt und sowohl für REM als auch TEM gebraucht.
Zur Untersuchung der Haemocyten ohne Zugabe eines Mediums wurde Haemolymphe in BEEM-Kapseln ( = 5,4 mm, pyramidenförmige Spitze; BALTEC) gegeben und nach 10 min, 30 min oder sofort in den Kapseln fixiert und für EM vorbereitet. |
| Graces-M. | 'WENNING-Ringer' | Kalium-Ring | |
|---|---|---|---|
| Graces Komp. I | |||
| Graces Komp. II | |||
| NaHCO3 | |||
| NaCl | |||
| KCl | |||
| CaCl2 mit 2 H2O | |||
| MgCl2 mit 6 H2O | |||
| Trehalose • 2 H2O | |||
| Hepes | |||
| pH | |||
| mit H2O auffüllen auf |
| Alle toten Partikel wurden vor ihrem Gebrauch im Versuch
mehrmals durch Suspendieren in a. demin. und Abzentrifugieren gewaschen
und schließlich in WENNING-Ringer oder Graces-Medium resuspendiert.
Lebende Bakterien (Micrococcus luteus Ml 11, Escherichia coli K12 D31 und Enterobacter cloacae 12; die Stämme stellte freundlicherweise die Arbeitsgruppe von Prof. Dr. P. Götz, FU Berlin, zur Verfügung.) wurden auf Nähragar (Standard I; Serva) bei 4 °C gehalten, in Nährbouillon (Standard I; Merck) im Schüttelbad bei 37 °C über Nacht in Kultur genommen und am nächsten Morgen in einer Wachstumskultur auf eine optische Dichte von 0,65 ( = 546 nm) gebracht; nach mehrfachem Waschen in Haemocyten-Kulturmedium wurden 10 µl der Bakteriensuspension pro ml Medium eingesetzt. Als relativ inerte, unbelebte Partikel wurden Latexkugeln von 5,1 ± 0,072 µm (LB5) bzw. 15,8 ± 2,8 µm (LB16) Durchmesser, Cytodex 1 (ø= 153 ± 26 µm) und Baumwollfasern verwendet. |
Zymosan, fixierte Ratten-Erythrocyten (RRBC) und lyophilisierter
Micrococcus 'lysodeikticus' (lMl) wurden als weniger inerte, tote
Partikel benutzt (s. Anhang).
Zudem wurde Zymosan mit FITC gekoppelt, um es fluoreszenzmikroskopisch, nach Injektion in das Haemocoel, in Haemocyten erkennen zu können. Zur Herstellung von Zymosan-FITC wurden 10 mg Zymosan im Eppendorfgefäß durch mehrmaliges Ultraschallsuspendieren in 1 ml a. demin. und Abzentrifugieren gewaschen. Eine kleine Spatelspitze NaHCO3 wurde in der Suspension gelöst und erst danach ca. 1 mg FITC zugegeben und das Gemisch für 30 min bei 40 °C im Wärmeschrank inkubiert. FITC reagiert im Alkalischen z.B. mit freien Aminogruppen (s. Abb. 3), die sich in und auf dem Zymosan befinden, es wird dadurch kovalent gebunden. Überschüssiges FITC wird nach der Reaktion durch mehrmaliges Waschen in a. demin. entfernt. |
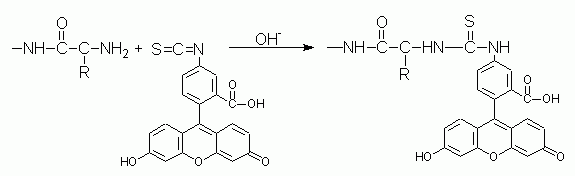
Abb. 3: Kopplung von FITC an frei Aminogruppe
| FITC-Zymosan wurden in Graces-Medium suspendiert, die Partikeldichte auf etwa 125.000/µl eingestellt und in der Bürkerkammer genau ausgezählt. Die Lebendgewichte der Versuchstiere wurden mit der Analysenwaage bestimmt, um den narkotisierten Tieren etwa 500.000 Partikel/100 mg Lebendgewicht in das | Haemocoel injizieren zu können. Zwei Stunden nach der Injektion wurde ihnen Haemolymphe entnommen und fluoreszenzmikroskopisch in der Bürkerkammer die Phagocytoserate bestimmt. |
| Zwei unterschiedliche in-vitro-Ansätze wurden erprobt: Einmal kamen die suspendierten Partikel zu Haemocytenkulturen in Blockschälchen auf Cellophan oder Glas und wurden für 13 h ohne Bewegung inkubiert und anschließend für EM fixiert. Im zweiten Fall kam Haemolymphe zu den im Kulturmedium suspendierten Partikeln in Eppendorfgefäßen. Diese Gefäße wurden dann an einem großen Magnetfloh mit Tesa®-Film befestigt und an einem auf die Seite gestellten Magnetrührer 1 h langsam über ihre | Hochachse gedreht, so daß Sedimentieren der Haemocyten und Partikel ausgeschlossen war und die Haemocyten mit den Partikeln frei im Medium interagieren konnten (Abb. 4). Danach wurde vorsichtig abzentrifugiert, der Überstand verworfen und das Pellet mit kaltem Fixativ resuspendiert. Die mit bloßem Auge erkennbaren Knötchen können aus dem Fixativ isoliert und für EM bearbeitet werden. |
 |
Abb. 4:. Für die bewegte Zellkultur auf die Seite gestellter Magnetrührer. Die Eppendorfgefäße (Pfeilspitzen) am Magnetfloh enthalten 1 ml Kulturmedium mit z.B. LB16-Partikeln und 10 µl Haemolymphe |
Copyright © 1996
Dr. L. Nevermann
[ Letzte Aktualisierung 31.07.00]