Teil II
[Literatur]
[ vorherige Seite |
Index | nächste Seite ]
Für die EM wurden die Objekte mit 2,5 % Glutaraldehyd und 2 % Paraformaldehyd in 0,1 M Natrium-Cacodylat-Puffer pH 7,0 (abgewandelt nach KARNOVSKY 1965) 12 h fixiert und danach zweimal 15 min mit dem gleichen Puffer gespült. Mit 2 % OsO4 in Cacodylat-Puffer wurde 1 h nachfixiert, anschließend mit dem gleichen Puffer 30 min gewaschen und in einer aufsteigenden Acetonserie entwässert.
TEM-Präparate wurden in Araldit® M nach GLAUERT & GLAUERT (Herstellerangabe) eingebettet. 104 ml Araldit® werden dazu mit 96 ml Dodecylbernsteinsäure und 2 % Beschleuniger (DY 064) gemischt (ohne den Beschleuniger kann das Gemisch im Kühlschrank längere Zeit gelagert werden). Die Proben werden in Stufen (1:3, 1:1, 3:1, je 1 h) von Aceton in unverdünntes Araldit®-Gemisch überführt, dieses nach mindestens 6 h noch einmal gewechselt, dann zunächst 5 h bei 45 °C, danach 24 h bei 65 °C polymerisiert.
Ultradünnschnitte wurden mit einem Reichert OM U3 und Diamantmesser (Diatom) angefertigt und mit Formvar beschichteten Kupfernetzchen (75 Maschen/inch) aufgenommen. Die Kontrastierung erfolgte mit 1 % Uranylacetat (20 min) nach WATSON (1958) und mit Bleizitrat (35 min) nach REYNOLDS (1963). Semidünnschnitte für die Lichtmikroskopie wurden mit Glasmessern geschnitten und mit Toluidinblau gefärbt.
REM-Präparate wurden ohne weitere Intermedien im Aceton in die Kammer eines Kritischpunkttrockners (CD 030, BAL-TEC) gebracht und nach Austausch des Acetons gegen CO2 kritischpunktgetrocknet. Direkt danach wurden die Proben mit beidseitiger Klebefolie auf Objekhaltern befestigt und in einem "sputter coater" (E 5000, Polaron) in Argonatmosphäre bei 1,2 kV und 40 mA etwa 50 nm dick mit Gold beschichtet
Die TEM-Präparate wurden mit einem Zeiss EM 9 A, die REM-Präparate mit einem Joel SEM 1 untersucht. Fotografiert wurde am EM 9 A auf Planfilm 7 × 7 (Agfa ScientiaTM) bzw. am Joel SEM 1 auf Rollfilm 7 × 5 (Ilford FP4 120, ISO 25/22°) Negativmaterial.
Eine Reihe von Farbstoffen ist geeignet, bestimmte zelluläre Strukturen fluoreszenzmikroskopisch sichtbar zu machen, ohne die lebenden Zellen wesentlich zu beeinträchtigen. Im folgenden sollen die in dieser Arbeit eingesetzten Farbstoffe kurz beschrieben werden.
|
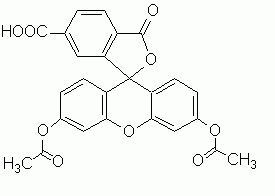
CFDA (6-Carboxyfluorescein di-Acetat) ist ein nichtpolarer, nicht fluorezierender Stoff, der sehr leicht in die Zellen diffundiert. Im Cytoplasma spalten Esterasen die Acetatreste ab, so daß ein polarer Fluoreszenzfarbstoff (CF, Carboxyfluorescein) entsteht, der die Zellen durch intakte Membranen nicht mehr verlassen kann und kaum in das Endosomale Kompartiment transportiert wird. Nach kurzer Färbezeit werden die Zellen gewaschen. Als Test auf die Ausbildung von 'Gap junctions' werden ungefärbte Zellen zu den gefärbten gegeben. Ein gutes Indiz für das Vorhandensein von Gap junctions zwischen den Zellen ist der Farbstofftransfer in die ungefärbten Zellen (CHURCHILL et al. 1993; GOODALL & JOHANSON 1982). |
|
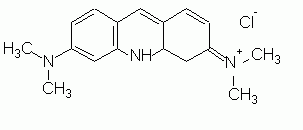
Acridin Orange (AO) ist membranpermeabel, bis es in ein saures Kompartiment gelangt, dissoziert und dann Membranen nicht mehr gut durchdringen kann (KNAPP & SWANSON 1990). AO wird daher in Lysosomen akkumuliert, wobei mit zunehmender Konzentration das emittierte Fluoreszenzlicht von Grün nach Orange-Rot wechselt (HART & YOUNG 1975; MATTEONI & KREIS 1987) Die Fluoreszenzfarbe ist aber auch pH-abhängig. Durch die Akkumulation kann AO osmotisch wirksam werden und zu einer Deformation der Lysosomen führen (KNAPP & SWANSON 1990). Nach kurzer Färbezeit (ca. 10 min) werden die Zellen gewaschen, die gefärbten Organellen halten aber den Farbstoff mehrere Tage. |
 |
Lucifer Yellow (LY) ist gut wasserlöslich und nicht
membranpermeabel. Der Farbstoff wird von Zellen bei Pinocytosevorgängen
ohne Adsorption proportional zur Konzentration im Kulturmedium mit diesem
zusammen aufgenommen (SWANSON 1989). Pinosomen, Endosomen und nach längerer
Inkubation auch Lysosomen werden angefärbt. In Lysosomen kann der Farbstoff
durch Transportprozesse konzentriert werden. Im Gegensatz zu AO soll LY keinen
Einfluß auf Form und Beweglichkeit von Lysosomen haben (KNAPP &
SWANSON 1990).
|
In der Anwendung sehr ähnlich wie LY ist FITC-Dextran, welches aufgrund verschiedener Dextrangruppen in diversen Molekulargewichten erhältlich ist. LY und FITC-Dextran werden hochkonzentriert (0,5 bis 10 mg/ml) zu den Zellkulturen gegeben und zeigen auch über mehrere Tage hinweg keine erkennbare Giftwirkung.
Mit FITC, TRITC oder Texas red gekoppelte Proteine wie Peroxidase (Px) oder Rinderalbumin eignen sich ebenfalls zur Markierung von Endosomen. Allerdings ist hier eine stärkere Aufnahme durch Adsorption an der Zelloberfläche wahrscheinlich; auch ist es möglich, daß sie enzymatisch hydrolysiert werden. Dadurch können die Fluorophoren innerhalb der Zelle eine andere Verteilung zeigen, als sie z.B. mit LY beobachtet wird. Die mit Rhodaminen (TRITC oder Texas red) gekoppelten Makromoleküle können eine verstärkte Adsorption auf der Zelloberfläche und damit auch eine verstärkte Aufnahme in die Zelle bewirken (SWANSON 1989).
Zelleigene Moleküle oder Rezeptoren lassen sich mit an Fluorophoren gekoppelten Stoffen darstellen, die mit recht hoher Spezifität ein bestimmtes Molekül binden oder von einem bestimmten Rezeptor gebunden werden. Lektine können bestimmte Zuckermoleküle nachweisen. Aufgrund der Zuckerspezifität für N-Acetylneuraninsäure (Sialinsäure, NANA, NeuNAc) und NAcetylDGlucose (DglcNAc Monomer und das 1-4 Dimer) gilt WGA (Weizenkeim-Agglutinin) als Marker für den Golgi-Komplex (TARTAKOFF & VASSALLI 1983).
Um einen möglicherweise vorhandenen Rezeptor für Lipopolysaccharid (LPS, ein Bestandteil der Bakterienzellwand) auf den Haemocyten darzustellen, wurden die Zellen mit LPS-FITC inkubiert.
Die Färbelösungen wurden mit Stammlösungen der Farbstoffe (Tab. 2) direkt vor den Färbungen angesetzt. Die Haemocyten wurden bis zu 30 min in Graces-Medium vorgespreitet, dann das Medium vorsichtig abgesaugt und durch 200 µl Färbelösung ersetzt. Nach den in Tab. 2 angegebenen Inkubationszeiten in der abgedunkelten Feuchten Kammer wurde vorsichtig mit Graces-Medium gespült.
Bei den mit CFDA gefärbten Präparaten wurde das Kulturmedium nach der Inkubationszeit zehnmal gewechselt, um möglichst jeden Farbstoffrest auszuschwemmen. Zu so gefärbten Haemocyten wurde frische Haemolymphe getropft und sofort ein Deckglas aufgelegt. Diese CF-Präparate wurden in zeitlichen Abständen von ca. 5 min jeweils unter Fluoreszenz- und Phasenkontrastbedingungen fotografiert. Um phototoxische Effekte zu minimieren wurde mit zusätzlichen Grün- und Graufiltern im Beleuchtungsstrahlengang mikroskopiert und das Filmmaterial auf 3200 ASA belichtet.
Tab. 2: Floureszenzfarbstoffe
| Stoff bzw. Konjugate | Stammlösung | Gebrauchslösung in Graces M. |
Inkubationszeit mit Farbstoff |
|---|---|---|---|
| AO | 2 mg/ml Graces-M |
5 µg/ml |
5 - 20 min |
| CFDA | 50 mg/ml DMSO |
50 µg/ml |
10 min |
| FITC-Dextran | 20 mg/ml DBS |
2 u. 5 mg/ml |
15 min - 3 h |
| FITC-Px | 2 mg/ml a. bidest. |
0,3 mg/ml |
15 min - 3 h |
| TRITC-Albumin | 1,25 mg/ml DBS |
0,1 u. 0,5 mg/ml |
15 min - 3 h |
| Texas red-Albumin | 1,25 mg/ml DBS |
0,1 u. 0,5 mg/ml |
15 min - 3 h |
| LY | 5 mg/ml Graces-M. |
2 mg/ml |
60 min - 3 h |
| WGA-FITC | 1 mg/ml DBS |
10 µg/ml |
15 - 30 min |
| LPS-FITC | 0,25 mg/ml H2O |
10 µg/ml |
30 - 60 min |
WGA-FITC und LPS-FITC fanden auch für fixierte Haemocyten Verwendung: nach 45 min Spreiten wurden die Zellen mit eiskaltem Methanol 10 min überschichtet, dann mit Graces-Medium gespült und mit der gleichen Färbelösung behandelt wie die lebenden Zellen.
Eingedeckelt wurde in allen Fällen mit Graces-Medium und einem mit
Siliconschliffett umrandetem Deckglas.
Auf Objektträgern gespreitete Haemocyten (s. Kap. 2.3.2) wurden mit 2,5 % Glutaraldehyd (in 0,01 M Cacodylat-Puffer, pH 7) 30 - 60 min in der Feuchten Kammer fixiert, dann mit Cacodylat-Puffer gespült.
Die Prophenoloxidase (ProPO) läßt sich mit Ethanol, anderen organischen Lösungsmitteln und SDS unphysiologisch aktivieren (PRESTON & TAYLOR 1970; ASHIDA & YAMAZAKI 1990). Um die größte Aktivität zu erhalten, wurde ein Teil der Präparate vor der Inkubation mit Substrat 15 min mit unvergälltem 30%igem Ethanol/Puffer aktiviert (vergällter Alkohol kann zu einer Hemmung der Enzymaktivität führen). Anschließend wurde mit Puffer gespült und 3mal 5 min überschichtet.
Als Substrat wurden unter Erwärmen 100 mg Dopa in 25 ml a. demin. gelöst, erst dann 0,05 g Na-Cacodylat zugegeben (BOGUSCH 1990) und nach Abkühlen der Lösung auf RT der pH-Wert mit HCl auf 7,0 eingestellt. Die Lösung unterliegt Autooxidation; daher wurde mit Stickstoff begast oder mit der Wasserstrahlpumpe entgast. Bei -18 °C eingefroren, kann der Substratansatz für mehrere Tage aufbewahrt werden.
Mit der Dopa-Lösung wurden die Haemocyten-Präparate überschichtet und für unterschiedliche Zeit inkubiert: Lithobius-Haemocyten 40 min bis 20 h, Scolopendra-Haemocyten über Nacht (16-20 h).
EM-Präparate wurden nach der aldehydischen Fixierung mit Cacodylatpuffer gespült und vor der Osmium-Fixierung 6 h (L. forficatus) oder 8 h (S. cingulata) mit der Dopa-Lösung inkubiert.
Als Negativkontrolle wurde mit PTU (Phenylthioharnstoff; wenige kleine Kristalle pro Objekträger) inhibiert und/oder die Ethanolaktivierung ausgelassen. Die fertigen Präparate wurden mit Wasser gespült und naß oder eingedeckelt in KAISERS Glyceringelatine lichtmikroskpisch untersucht
Die Fixierung der gespreiteten Haemocyten erfolgte wie in 2.5.2.1 beschrieben. Anschließend wurden die Zellen mit Tris/HCl-Puffer (0,05 M, pH 7,6) gespült und durch dreimaliges Überschichten jeweils 5 min gewaschen. In der abgedunkelten Feuchten Kammer wurden die Haemocyten 30 min bis 1 h mit Substratlösung (10 mg DAB und 6 µl 30%iges H2O2 in 20 ml 0,05 M Tris/HCl, pH 7,6; s. ROMEIS 1989) inkubiert; durch Spülen mit Wasser wurde die Reaktion gestoppt. Zur Kontrolle wurden parallel dazu Haemocyten in Substratlösung ohne H2O2 oder in Substratlösung mit Aminotriazol (0,02 M in Tris/HCl) 1:1 oder mit Substratlösung und ein paar Kristallen PTU inkubiert. Eingedeckelt wurde mit KAISERS Glyceringelatine.
Der Nachweis erfolgte durch direkte Azokupplung mit stabilem Diazoniumsalz (s. ROMEIS 1989): Als Substrat wurden 416 mg Naphtol AS-TR Phosphat zunächst in 0,5 ml Dimethylformamid gelöst und dann mit 20 ml Natriumacetatpuffer (0,1 M, pH 5,0 oder 6,0) aufgefüllt; 510 mg Fast Blue BB Salz wurden kurz vor der Färbung darin gelöst. Als Inhibitor diente 0,1 M NaF im Reaktionsgemisch. Gespreitete Haemocyten wurden mit 3,5%igem Formaldehyd in 0,1 M Phosphatpuffer pH 7,2 fixiert und anschließend 1 min mit 0,5 % (v/v) Triton X-100 in Acetatpuffer inkubiert, danach mit dem gleichen Puffer gespült und 3mal 5 min überschichtet, am Ende mit filtrierter Substratlösung bis zu 1 h überschichtet. Nach dem Spülen mit Wasser wurde in KAISERS Glyceringelatine eingedeckelt.
Der Nachweis erfolgt durch direkte Azokupplung (s. ROMEIS 1989). Es wurden 5 mg Naphtol AS-BI Phosphat in 0,5 ml Dimethylformamid gelöst und in 10 ml 0,1 M Tris/HCl-Puffer pH 9,0 überführt; 10 mg Fast Blue BB Salz wurden erst kurz vor der Färbung zugegeben. Gespreitete Haemocyten wurden mit 3,5%igem Formaldehyd in 0,1 M Tris/HCl-Puffer pH 9,0 fixiert, mit dem Tris/HCl-Puffer gewaschen und 3 mal 5 min überschichtet, dann bis zu 2 h mit filtrierter Substratlösung inkubiert. Nach dem Spülen mit Wasser wurde in KAISERS Glyceringelatine eingedeckelt.
Der Nachweis erfolgt durch direkte Azokupplung (verändert nach BURCK 1982). Es wurden 5 mg Naphtol AS Acetat in 50 µl Aceton gelöst und mit 20 ml PBS (pH 6,5) aufgefüllt. Direkt vor der Färbung wurden 5 mg Fast Garnet GBC in 50 µl Aceton suspendiert und zu der Naphtol AS Acetat-Lösung gegeben, kräftig gemischt, gleich auf die Präparate filtriert und 1 h inkubiert. Gespreitete Haemocyten wurden vor der Inkubation mit 3,5%igem Formaldehyd in 0,2 M Phosphatpuffer (pH 6,5) fixiert, 1 min mit 0,5 % (v/v) Triton X100 inkubiert und mit PBS gespült. Nach der Färbung wurde mit a. demin. gespült und in KAISERS Glyceringelatine eingedeckelt.
Mit einer einfach-indirekten immuncytochemischen Methode wurde lysozymartige Immunreaktivität auf Spreitungspräparaten nachgewiesen. Als Primärantikörper wurde Anti-Human-Lysozym-Antikörper (Anti-Lysozym-AK) vom Kaninchen und als Sekundärantikörper Anti-Kaninchen-AK gekoppelt mit alkalischer Phosphatase (aPh) eingesetzt. Zur Lokalisation der aPh wurde Sigma Fast Fast Red TR/Naphtol AS-MX oder Sigma Fast BCIP/NBT als Substrat benutzt, die rote bzw. graublaue, wasserunlösliche Produkte liefern.
Gespreitete Haemocyten wurden in 0,2 % Glutaraldehyd und 4 % Formaldehyd in 0,05 M Na-Cacodylatpuffer (pH 7,0) 30-60 min fixiert und mehrmals mit TBST (s. Anhang für Abkürzungen) gespült. Unspezifische Bindungsstellen wurden durch überschichten mit BSA-TBST (30 min) blockiert.
Mit dem Primärantikörper gegen Lysozym (1:500 und 1:1000 in BSA-TBST) wurde 2½ h bei RT oder über Nacht bei 4 °C inkubiert. Nicht gebundene Anti-Lysozym-AK wurden durch 6mal 5minütiges Überschichten mit BSA-TBST gründlich entfernt und die Objekte mit Sekundärantikörper (1:200 und 1:400 in BSA-TBST) 2 h inkubiert. Auch der Sekundärantikörper wurde gründlich mit TBST abgespült, bevor die Substratlösung für die Färbereaktion auf die Präparate kam.
Als Kontrollen wurden entsprechende Spreitungspräparate jeweils ohne Primärantikörper bzw. ohne Sekundärantikörper der Färbereaktion unterzogen. Durch Spülen mit a. demin. wurden die Färbereaktion gestoppt und die Präparate anschließend in KAISERS Glyceringelatine eingedeckelt.
Für die lichtmikroskopischen Untersuchungen und zur Dokumentation wurden benutzt:
Diapositive von Semidünnschnitten wurden auf Papier projiziert und darauf die Konturen der abgebildeten Haemocyten und ihrer Kerne nachgezeichnet. Die umrandeten Flächen wurden ausgeschnitten und gewogen. Aus den relativen Gewichten wurden Anschnitt-Flächenverhältnisse und Kern-Plasma-Verhältnisse berechnet und dabei auch solche Zellanschnitte berücksichtigt, die keinen Kern enthielten, so daß die Flächenverhältnisse den Volumenverhältnissen entsprechen sollten. Bei der Berechnung von Flächenverhältnissen der Zelltypen zueinander wurden die Vergrößerungen der Einzelbilder berücksichtigt, bei der Berechnung der Kern-Plasma-Verhältnisse die Häufigkeit der einzelnen Zelltypen im jeweiligen Einzelbild (s. Anhang).
Die Lysozymaktivität wurde nach dem Prinzip von CERIOTTI (MOHRIG & MESSNER 1968) in einem Agar-Diffusions-Lysishoftest quantifiziert. Dazu wurde ein 1%iger Oxoid®-Agar mit 200 mg gewaschenem Ml pro 100 ml in DBS (s. Anhang) angesetzt und autoklaviert. Von diesem Ml-Agar wurden je 7 ml in Petrischalen (ø = 10 cm) verteilt und nach dem Erkalten mit einer Hülse (ø = 3 mm) ein symmetrisches Neun-Loch-Muster in den Agar gestanzt. In die Löcher wurden 4 µl Probe oder Standard pipettiert. Als Standard dienten 6,5 mg Hühner-Eiklar-Lysozym pro 10 ml DBS und eine Verdünnungsreihe davon: entsprechend wurden 2,6; 1,3; 0,13; 0,065; 0,013 µg Standard-Lysozym pro Loch einpipettiert. Lithobius-Haemolymphe wurde 1:1 mit 1/2, 1/250 u. 1/500 Anti-Lysozym-AK/DBS und 1:1 mit DBS verdünnt auf den Platten getestet.
Mit Parafilm verschlossen wurden die Petrischalen 24 h im Wärmeschrank bei 30 °C inkubiert, entstandene Lysishöfe mit einer Schieblehre ausgemessen.
Zur elektrophoretischen Auftrennung von Haemolympheproteinen diente eine Mini-Twin "Slabgelapparatur" von Biometra® und ein regelbarer Konstantstrom-Transformator von Sebia.
Polyacrylamidgele verschiedener Konzentration (7,5; 10; 15; 20 %) und lineare Gradienten-Gele (3 o. 5% bis 10; 15 o. 20 %), jeweils mit Sammelgelen von 3 oder 3,75 %, wurden unter Verwendung des diskontinuierlichen Puffersystems von LAEMMLI (1970) in den Minigelplatten (8,6 × 7,7 × 0,1 cm3) hergestellt.
Die Gradientengele wurden mit Hilfe eines Gradientenmischers (Eigenbau) gegossen. Die Ansätze für die 'leichten' 3- bzw. 5%igen und die 'schweren' 10-, 15- bzw. 20%igen Komponenten sind in Tab. 3, die für die Sammelgele in Tab. 4 dargestellt.
Im einzelnen wurden eingesetzt (Endkonzentrationen, Stammlsg. in Klammern):
Tab. 3: Ansätze für je 12 ml Grandientengelkomponenten.
| Gelkonzentration | |||||
|---|---|---|---|---|---|
3 % |
5 % |
10 % |
15 % |
20 % |
|
Acrylamidlsg. |
1,2 ml |
2 ml |
4 ml |
6 ml |
8 ml |
Trennpuffer |
3 ml |
3 ml |
3 ml |
3 ml |
3 ml |
Succrose |
- |
- |
1,8 g |
1,8 g |
1,8 g |
H2O |
7,8 ml |
7 ml |
4 ml |
2 ml |
- |
APS |
15 µl |
15 µl |
15 µl |
15 µl |
15 µl |
Entgasen |
+ |
+ |
+ |
+ |
+ |
davon je |
3 ml |
3 ml |
3 ml |
3 ml |
3 ml |
und TEMED |
15 µl |
15 µl |
15 µl |
15 µl |
15 µl |
Tab. 4: Ansätze für Sammelgele
Sammelgelkonz. |
||
|---|---|---|
3 % (10 ml) |
3,75 % (8 ml) |
|
Acrylamidlsg. |
6,5 ml |
5 ml |
Sammmelgelpuffer |
1 ml |
1 ml |
H2O |
2,5 ml |
2 ml |
APS |
75 µl |
60 µl |
Entgasen |
+ |
+ |
TEMED |
610 µl |
610 µl |
Frisch entnommene oder eingefrorene Haemolymphe wurde mit doppelt konzentriertem Probenpuffer (ohne reduzierende Stoffe wie Mercaptoethanol oder Dithiothreitol) 1:1 gemischt und für wenige Sekunden mit 40 W an einem Branson-Sonifier B-12 homogenisiert. Die Proben wurden dabei und auch später nicht erhitzt, um Enzyme nicht irreversibel zu denaturieren.
Neben den Proben (510 µl/Spur) wurden Molekulargewichtsmarker (SDS-6, SDS-7B o. SDS-K, s. Chemikalienliste im Anhang) aufgetragen.
Für die Elektrophorese wurde die Apparatur in einem Kühlschrank (4 °C) untergebracht, und der Konstantstrom in Stufen (1.-5. min 10 mA, 5. bis 10. min 15 mA, restliche Zeit 20 mA pro Gel) geschaltet.
Nach der Elektrophorese wurden die Gele für den Enzymnachweis oder Western Blot weiter bearbeitet oder mit 10 % Methanol, 7,4 % Essigsäure und Spuren von Coomassie-Blau über Nacht gefärbt. Zum Aufbewahren wurden die Gele in einem Methanol-Glycerin-Wasser-(27:3:70)-Gemisch imprägniert und in Spannrahmen zwischen Cellophan getrocknet.
Der Nachweis von PO- und Px-Aktivität erfolgte verändert nach NELLAIAPPAN & VINAYAKAM (1993) und NELLAIAPPAN & VALIVITTAN (1993). Im Anschluß an die SDS-PAGE wurden die Trenngele 3mal 15 min in 2 % (v/v) Triton X100 in a. demin. und 3mal 15 min in Cacodylat-Puffer (0,01 M, pH 7) bzw. in TBS geschwenkt, um das SDS auszuwaschen und die Proteine zu renaturieren. Für die Färbreaktionen dienten die in den Kapiteln 2.5.2.1 und 2.5.2.2 beschriebenen Substratlösungen; durch Spülen der Gele in a. demin. wurde die Reaktion gestoppt. Weiterhin wurden Gele nach dem Spülen mit Triton X100 mit 20 mM Tropolon in Cacodylat-Puffer und anschließend mit Dopa und Tropolon inkubiert. Tropolon gilt als Inhibitor der PO, kann aber Substrat für die Px sein (KAHN 1985).
Um festzustellen, ob die Lysozymaktivität vorwiegend in den Haemocyten oder im Haemolympheplasma vorhanden ist, mußten Haemocyten- und Plasmaproteine voneinander getrennt werden.
Dazu wurde Haemolymphe von der Wunde mit silikonisierten Kapillaren aufgenommen, sofort mindestens 1:20 mit eisgekühltem Antikoagulationspuffer (50 mM tri-Na-Citrat, 10 mM EDTA, 31 mM NaCl, 0,5 mM PMFS, pH 6,5) verdünnt und anschließend bei 100 ×g kurz abzentrifugiert. Plasma/Puffer-Überstand und Haemocyten-Pellet wurden getrennt weiter verarbeitet:
Die eingeengten Überstände werden jeweils auf den Filtern mit Probenpuffer (mit EDTA und PMFS) vermengt und aufgenommen.
In Anlehnung an die Lysishoftests und in Abwandlung nach AUDY et al. (1989) wurden zum Nachweis einer Lysozymaktivität 2,5 mg lyophilisierter Micrococcus 'lysodeikticus' (Ml) pro ml Gelansatz suspendiert und im Gel einpolymerisiert (Ml wurde zuvor in a. demin. ultraschallsuspendiert und durch Zentrifugation und Resuspension 3mal gewaschen). Nach der Elektrophorese wurden die Trenngele in 2 % (v/v) Triton® X100 in a. demin. geschwenkt, bis Lysishöfe in den milchig-trüben Gelen sichtbar wurden (30-60 min; eine Zugabe von etwas DBS zu der Tritonlösung scheint die Entwicklung der Lysishöfe zu beschleunigen).
Die Ml-Gele wurden nicht getrocknet, da sie beim Trocknen klar werden und die Lysishöfe dann nicht mehr erkennbar sind (durch erneutes Quellen der Gele in Imprägnierlösung erscheinen sie allerdings wieder).
Haemolympheproben (in doppelt konzentriertem Probenpuffer mit oder ohne EDTA, PMFS; s.o.) und Molekularstandard (SDS-K) werden auf 15%igen Ml-PA-Gelen so nebeneinander aufgetragen, daß das Gel später in zwei spiegelsymmetrische Hälften geteilt werden kann. Nach der Elektrophorese dient eine Hälfte zur Kontrolle der Lysozymaktivität (s. Kap. 2.10), die andere wird für den Western Blot benutzt.
Als 'Semidry-Blot-Kammer' dient ein Eigenbau mit Edelstahlelektroden auf Kunststoffträgerplatten mit integrierter Wasserkühlung. Die Anode wird durch eine plangeschliffene Schicht "Leit-C nach Göcke" (Plano) gegen Elektroerosion geschützt (Abb. 5).
Geblottet wurde auf Trans-Blot® (BioRad) Nitrocellulosemembranen
(Porengröße 0,2 µm). Kontakt zum Gel und zur NC-Membran stellten
je 4 Lagen transferpuffer-getränktes Filterpapier her [Transferpuffer
nach TOWBIN et al. (1979): 25 mM Tris, 190 mM Glycin in 20 % Methanol pH
8,3]. Ein guter Transfer war (am Molekularstandard erkennbar) bei 1,5
mA/cm2 Gel nach 1,5 Stunden erreicht.
 |
Abb. 5: Semidryblotter 'UMRB' |
Die NC-Membran wurde danach kurz mit a. demin. gespült und mit oder ohne vorherige Fixierung (20 min, Fixativ für Immunhistochemie, s. Kap. 2.5.3) 30 min luftgetrocknet. Mögliche freie Protein-Bindungsstellen auf der Membran wurden danach 20 min mit 5 % Milchpulver und 1 % BSA in TBST behandelt.
Die Blotmembran wurde dann über Nacht in 1/1000 Anti-Lysozym-AK in BSA-TBST bei 4 °C gebadet, am nächsten Tag 2mal kurz mit TBST und 2mal 10 min mit BSA-TBST gewaschen, bevor 2 h mit 1/600 Anti-Kaninchen-IgG-aPh in BSA-TBST inkubiert wurde. Vor der Färbereaktion mit Sigma Fast® wurde 1mal kurz und 3mal 10 min in TBST gespült, nach der Färbung mit a. demin. gewaschen und auf Filterpapier getrocknet.
[Literatur]
[ vorherige Seite |
Index | nächste Seite ]
Copyright © 1996
Dr. L. Nevermann
[ Letzte Aktualisierung 29.07.00]